
Maladie polykystique des reins autosomique dominante - Autosomal dominant polycystic kidney disease
| Maladie polykystique des reins autosomique dominante | |
|---|---|
| Autres noms | PKD autosomique dominante, PKD de l'adulte |
 | |
| Reins polykystiques | |
| Spécialité |
Génétique médicale |
La maladie polykystique rénale autosomique dominante ( PKRAD ) est la maladie humaine monogénique potentiellement mortelle la plus répandue . Elle est associée à une grande variabilité interfamiliale et intrafamiliale, qui s'explique en grande partie par son hétérogénéité génétique et ses gènes modificateurs . C'est également la plus courante des maladies rénales kystiques héréditaires - un groupe de troubles avec une pathogenèse apparentée mais distincte, caractérisé par le développement de kystes rénaux et diverses manifestations extrarénales, qui en cas de PKRAD incluent des kystes dans d'autres organes, tels que le foie , les vésicules séminales , le pancréas et la membrane arachnoïdienne , ainsi que d'autres anomalies telles que les anévrismes intracrâniens et les dolichoectasies , la dilatation et les anévrismes de la racine aortique, le prolapsus de la valve mitrale et les hernies de la paroi abdominale . Plus de 50 % des patients atteints de PKRAD développent éventuellement une maladie rénale en phase terminale et nécessitent une dialyse ou une transplantation rénale . On estime que la PKRAD affecte au moins une personne sur 1000 dans le monde, faisant de cette maladie la maladie rénale héréditaire la plus courante avec une prévalence diagnostiquée de 1:2000 et une incidence de 1:3000-1:8000 à l'échelle mondiale.
Signes et symptômes
Parmi les présentations cliniques figurent :
- Douleur aiguë au rein
- Sang dans les urines
- Reins au ballottage
- Hémorragie sous-arachnoïdienne (anévrisme de la baie)
- Hypertension
- Kystes hépatiques associés
- Urémie due à une insuffisance rénale
- Anémie due à une maladie rénale chronique
- Augmenter la sécrétion de globules rouges ou d' érythropoïétine
La génétique
L'ADPKD est génétiquement hétérogène avec deux gènes identifiés : PKD1 (région chromosomique 16p13.3 ; environ 85 % des cas) et PKD2 (4q21 ; environ 15 % des cas). Plusieurs mécanismes génétiques contribuent probablement à l' expression phénotypique de la maladie. Bien qu'il existe des preuves d'un mécanisme à deux coups (inactivation germinale et somatique de deux allèles PKD) expliquant le développement focal des kystes rénaux et hépatiques, l' haploinsuffisance est plus susceptible d'expliquer les manifestations vasculaires de la maladie. De plus, de nouveaux modèles de souris homozygotes pour les allèles hypomorphes PKD1 22 et 23 et la démonstration d'une prolifération accrue des cellules épithéliales rénales chez les souris PKD2 +/- suggèrent que des mécanismes autres que l'hypothèse des deux coups contribuent également au phénotype kystique.
Une grande variabilité interfamiliale et intrafamiliale se produit dans la PKRAD. La plupart des individus porteurs de mutations PKD1 présentent une insuffisance rénale à l'âge de 70 ans, alors que plus de 50 % des individus porteurs de mutations PKD2 ont une fonction rénale adéquate à cet âge (âge moyen d'apparition de l'insuffisance rénale terminale : 54,3 ans avec PKD1 ; 74 ·0 ans avec PKD2 ).
La variabilité intrafamiliale significative observée dans la gravité des manifestations rénales et extrarénales indique des facteurs modificateurs génétiques et environnementaux qui peuvent influencer l'issue de la PKRAD, et les résultats d'une analyse de la variabilité de la fonction rénale entre les jumeaux monozygotes et les frères et sœurs soutiennent le rôle des modificateurs génétiques dans cette maladie. On estime que 43 à 78 % de la variance de l'âge par rapport à l'IRT pourrait être due à des facteurs modificateurs héréditaires, les parents étant aussi susceptibles que les enfants de présenter une maladie plus grave dans les études sur les paires parent-enfant.
Physiopathologie
Chez de nombreux patients atteints de PKRAD, la dysfonction rénale n'est cliniquement apparente qu'à 40 ou 50 ans. Cependant, de plus en plus de preuves suggèrent que la formation de kystes rénaux commence in utero . Les kystes se forment initialement sous forme de petites dilatations dans les tubules rénaux, qui se dilatent ensuite pour former des cavités remplies de liquide de différentes tailles. Les facteurs suggérés pour conduire à la cystogenèse comprennent une mutation germinale dans l'un des allèles du gène de la polycystine, un deuxième coup somatique qui conduit à la perte de l'allèle normal, et un troisième coup, qui peut être une atteinte rénale qui déclenche la prolifération cellulaire, et un réponse à la blessure. . En raison de nombreuses similitudes entre la physiopathologie de la PKRAD et la physiopathologie de la réponse rénale aux lésions, la PKRAD a été décrite comme un état d'activation aberrante et persistante des voies de réponse aux lésions rénales. Au cours de la progression de la maladie, la dilatation continue des tubules par l'augmentation de la prolifération cellulaire, de la sécrétion de liquide et de la séparation du tubule parental conduit à la formation de kystes.
La PKRAD, ainsi que de nombreuses autres maladies associées à des kystes rénaux, peuvent être classées dans une famille de maladies appelées ciliopathies . Les cellules épithéliales des tubules rénaux, y compris tous les segments du néphron et les canaux collecteurs (à l'exception des cellules intercalées) montrent la présence d'un seul cil apical primaire. La polycystine-1 , la protéine codée par le gène PKD1 , est présente sur ces cils et on pense qu'elle détecte le flux avec ses grands domaines extracellulaires, activant les canaux calciques associés à la polycystine-2 , le produit du gène PKD2 , à la suite de le cadre génétique de la PKRAD comme expliqué dans la sous-section génétique ci -dessus.
La prolifération des cellules épithéliales et la sécrétion de liquide qui conduisent à la cystogenèse sont deux caractéristiques de la PKRAD. Au cours des premiers stades de la cystogenèse, les kystes sont attachés à leurs tubules rénaux parentaux et un dérivé du filtrat glomérulaire pénètre dans les kystes. Une fois que ces kystes s'étendent à environ 2 mm de diamètre, le kyste se ferme de son tubule parental et après cela, le liquide ne peut entrer dans les kystes que par la sécrétion transépithéliale, qui à son tour est suggérée pour augmenter en raison des effets secondaires d'une concentration intracellulaire accrue de cyclique AMP (AMPc).
Cliniquement, l'augmentation insidieuse du nombre et de la taille des kystes rénaux se traduit par une augmentation progressive du volume rénal. Des études menées par des professionnels de la Mayo Clinic ont établi que le volume total du rein (TKV) dans une large cohorte de patients atteints de PKRAD était de 1060 ± 642 ml avec une augmentation moyenne de 204 ml sur trois ans, soit 5,27 % par an dans l'évolution naturelle de la maladie, parmi d'autres découvertes importantes et nouvelles qui ont été largement étudiées pour la première fois.

Diagnostic
Habituellement, le diagnostic de PKRAD est initialement réalisé par imagerie rénale à l'aide d'une échographie , d'une tomodensitométrie ou d'une IRM . Cependant, le diagnostic moléculaire peut être nécessaire dans les situations suivantes : 1- lorsqu'un diagnostic définitif est requis chez des individus jeunes, comme un donneur potentiel vivant apparenté dans une famille affectée avec des données d'imagerie équivoques ; 2- chez les patients ayant des antécédents familiaux négatifs de PKRAD, en raison d'un chevauchement phénotypique potentiel avec plusieurs autres maladies kystiques rénales ; 3- dans les familles touchées par une maladie polykystique des reins à début précoce, car dans ce cas des allèles hypomorphes et/ou héréditaires oligogéniques peuvent être impliqués ; et 4- chez les patients sollicitant un conseil génétique , notamment chez les couples souhaitant un diagnostic génétique préimplantatoire .
Les résultats de gros reins échogènes sans kystes macroscopiques distincts chez un nourrisson/enfant présentant un risque de 50 % de PKRAD sont diagnostiques. En l'absence d'antécédents familiaux de PKRAD, la présence d'une hypertrophie rénale bilatérale et de kystes, avec ou sans présence de kystes hépatiques , et l'absence d'autres manifestations suggérant une maladie kystique rénale différente fournissent des preuves présumées, mais non définitives, de le diagnostic. Dans certains cas, les anévrismes intracrâniens peuvent être un signe associé de PKRAD, et le dépistage peut être recommandé pour les patients ayant des antécédents familiaux d'anévrisme intracrânien.
Les tests de génétique moléculaire par analyse de liaison ou dépistage direct de mutations sont disponibles en clinique ; cependant, l'hétérogénéité génétique est une complication importante des tests de génétique moléculaire . Parfois, un nombre relativement important de membres de la famille touchés doit être testé afin d'établir lequel des deux gènes possibles est responsable au sein de chaque famille. La grande taille et la complexité des gènes PKD1 et PKD2 , ainsi que l' hétérogénéité allélique marquée , présentent des obstacles aux tests moléculaires par analyse directe de l' ADN . La sensibilité du test est de près de 100 % pour tous les patients atteints de PKRAD âgés de 30 ans ou plus et pour les patients plus jeunes présentant des mutations PKD1 ; ces critères ne sont sensibles qu'à 67 % pour les patients présentant des mutations PKD2 ] et âgés de moins de 30 ans.

Rein polykystique adulte

Diagramme de la maladie polykystique autosomique dominante avec un encart rénal normal pour comparaison
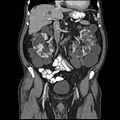
Tomodensitométrie abdominale d'un adulte atteint d'une maladie polykystique rénale autosomique dominante : une formation étendue de kystes est observée sur les deux reins, avec quelques kystes également dans le foie. ( Plan coronal )



Traitement
Actuellement, le seul traitement pharmacologique disponible pour la PKRAD consiste à réduire la vitesse de gain de volume rénal total (VTK) avec des antagonistes des récepteurs de la vasopressine 2 (V2) (c'est-à-dire le tolvaptan). Le traitement par le tolvaptan n'arrête ni n'inverse la progression de la maladie et les patients évoluent toujours vers une insuffisance rénale. Les modalités de traitement palliatif impliquent des médicaments symptomatiques (analgésiques non opioïdes et opioïdes) pour les douleurs abdominales/rétropéritonéales. Les options pour la douleur résistante aux analgésiques comprennent des interventions chirurgicales simples ou complexes (c.-à-d. aspiration de kyste rénal, décortication de kyste, dénervation rénale et néphrectomie), qui peuvent entraîner des complications inhérentes à la chirurgie. Des recherches récentes suggèrent que les interventions diététiques cétogènes affectent de manière bénéfique la progression et les symptômes chez les personnes atteintes de PKRAD. Une légère perte de poids affecte favorablement la douleur, indiquant le bénéfice des changements alimentaires et de style de vie.
Médicaments aquarétiques
En 2014, le Japon a été le premier pays au monde à approuver un traitement pharmacologique pour la PKRAD, suivi du Canada et de l'Europe, qui ont approuvé le médicament tolvaptan pour les patients atteints de PKRAD début 2015. La FDA américaine a approuvé l'utilisation du tolvaptan dans le traitement de la PKRAD. ADPKD en 2018. Le tolvaptan, un médicament aquarétique , est un antagoniste des récepteurs de la vasopressine 2 (V2) . Des études précliniques avaient suggéré que la molécule d' AMPc pourrait être impliquée dans l'élargissement des kystes de la PKRAD, et des études sur des rongeurs ont confirmé le rôle de la vasopressine dans l'augmentation des niveaux d'AMPc dans le rein, ce qui a jeté les bases de la conduite d'études cliniques. Parce que les données du Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) dirigé par la Mayo Clinic ont montré que le volume total du rein (TKV) prédisait le risque de développer une maladie rénale chronique chez les patients atteints de PKRAD, l'essai TEMPO 3:4, qui a inclus patients de 129 sites dans le monde de 2007 à 2009, ont évalué le TKV comme critère d' évaluation principal pour tester l'efficacité du tolvaptan chez les patients atteints de PKRAD. Cette étude a montré une diminution significative du rapport entre l'augmentation du TKV et l'effet dissuasif sur le déclin de la fonction rénale chez les patients atteints de PKRAD après un traitement par le tolvaptan ; cependant, étant donné que les résultats des tests de laboratoire concernant la fonction hépatique semblaient élevés chez un pourcentage de patients inclus dans cette étude, l'approbation du médicament a été soit retardée par les organismes de réglementation, soit, comme dans le cas des États-Unis, totalement refusée.
Interventions diététiques et de style de vie
Des recherches utilisant des modèles murins de PKRAD ont montré qu'une restriction alimentaire modérée améliorait fortement la progression de la maladie. Il a été démontré que le mécanisme impliquait l'état métabolique de la cétose, et des effets bénéfiques pourraient être produits par une alimentation limitée dans le temps, un jeûne aigu, un régime cétogène ou par une supplémentation en bêta-hydroxybutyrate de cétone dans des modèles murins, rats et chats de PKRAD. Un régime cétogène a non seulement stoppé la progression de la maladie, mais a conduit à une inversion partielle de la maladie kystique rénale dans un modèle de rat. L'état métabolique de la cétose peut être bénéfique dans la PKRAD car les cellules du kyste rénal dans la PKRAD ont un défaut métabolique similaire à celui du cancer à effet Warburg qui les rend fortement dépendantes du glucose et incapables de métaboliser les acides gras et les cétones. Conformément à cela, les niveaux de glucose sérique sont en corrélation positive avec une progression plus rapide de la maladie chez les patients atteints de PKRAD. En outre, les personnes atteintes de PKRAD et de diabète de type 2 ont un volume rénal total (TKV) significativement plus important que celles atteintes de PKRAD seule, et le surpoids ou l'obésité s'associent à une progression plus rapide de la PKRAD à un stade précoce. Une étude de série de cas rétrospective a montré que les symptômes de la PKRAD - y compris la douleur, l'hypertension et la fonction rénale - se sont améliorés chez 131 patients qui ont suivi un régime cétogène pendant une durée moyenne de 6 mois.
L'apport alimentaire en sodium est associé à une détérioration de la fonction rénale dans la PKRAD, et il est généralement recommandé de limiter l'apport en sodium aux patients. L'apport en protéines alimentaires n'a pas été trouvé en corrélation avec la progression de la PKRAD.
On pense que l'augmentation de la consommation d'eau est bénéfique dans la PKRAD et est généralement recommandée. Le mécanisme bénéfique sous-jacent de l'augmentation de la consommation d'eau peut être lié à des effets sur le récepteur de la vasopressine V2 ou peut être dû à la suppression de la formation de microcristaux nocifs dans les tubules rénaux par dilution de solutés tels que l'oxalate de calcium, le phosphate de calcium et l'acide urique.
Médicament analgésique
La douleur chronique chez les patients atteints de PKRAD est souvent réfractaire aux traitements conservateurs non invasifs, mais les analgésiques non opioïdes et les interventions conservatrices peuvent être d'abord utilisés avant d' envisager les analgésiques opioïdes ; si la douleur persiste, les interventions chirurgicales peuvent cibler les kystes rénaux ou hépatiques pour traiter directement la cause de la douleur, avec des options chirurgicales comprenant la décortication des kystes rénaux, la dénervation rénale et la néphrectomie .
Aspiration d'un kyste rénal
L'aspiration avec sclérothérapie à l' éthanol peut être réalisée pour le traitement des kystes rénaux simples symptomatiques, mais peut être peu pratique chez les patients avancés présentant des kystes multiples. L'intervention elle-même consiste en l'insertion percutanée d'une aiguille dans le kyste identifié, sous guidage échographique, suivie d'un drainage du liquide contenu ; la sclérothérapie est utilisée pour éviter la réaccumulation de liquide qui peut se produire dans le kyste, ce qui peut entraîner une récurrence des symptômes.
Décortication du kyste laparoscopique
La décortication laparoscopique des kystes (également appelée marsupialisation) consiste en l'ablation d'un ou plusieurs kystes rénaux par chirurgie laparoscopique , au cours de laquelle les kystes sont perforés, et la paroi externe des plus gros kystes est excisée en prenant soin de ne pas inciser le parenchyme rénal. Cette procédure peut être utile pour soulager la douleur chez les patients atteints de PKRAD et est généralement indiquée après qu'une aspiration antérieure du kyste a confirmé que le kyste à décortiquer est responsable de la douleur. Des essais contrôlés non randomisés menés dans les années 90 ont montré que les patients atteints de kystes rénaux simples symptomatiques qui présentaient une récurrence des symptômes après une réponse initiale à une simple aspiration pouvaient être soumis en toute sécurité à une décortication du kyste, avec une durée de vie moyenne sans douleur entre 17 et 24 mois après la chirurgie. La décortication laparoscopique présente un taux de récidive de 5 % des kystes rénaux par rapport à un taux de récidive de 82 % obtenu avec la sclérothérapie.
Neurolyse
Un nouveau traitement spécifique de la douleur chronique dont souffrent de nombreuses personnes atteintes de PKRAD est la neurolyse du plexus cœliaque . Cela implique l'ablation chimique du plexus cœliaque , pour provoquer une dégénérescence temporaire des fibres nerveuses ciblées. Lorsque les fibres nerveuses dégénèrent, cela provoque une interruption de la transmission des signaux nerveux. Ce traitement, lorsqu'il est réussi, procure un soulagement important de la douleur pendant une période allant de quelques jours à plus d'un an. La procédure peut être répétée lorsque les nerfs affectés sont guéris et que la douleur revient.
Néphrectomie
De nombreux patients atteints de PKRAD souffrent de séquelles symptomatiques de la maladie, telles qu'une hémorragie kystique , des douleurs au flanc , des infections récurrentes , une lithiase rénale et des symptômes d'effet de masse (c'est-à-dire une satiété précoce , des nausées et des vomissements et une gêne abdominale), de leurs reins hypertrophiés. Dans de tels cas, une néphrectomie peut être nécessaire en raison de symptômes insolubles ou lorsque, au cours de la préparation d' une transplantation rénale , les reins natifs empiètent sur le vrai bassin et empêchent la mise en place d'une allogreffe de donneur . De plus, une néphrectomie native peut être entreprise en présence d'une suspicion de malignité, car le carcinome à cellules rénales (RCC) est deux à trois fois plus probable dans la population PKRAD en phase terminale d'insuffisance rénale (ESKD) que chez les patients ESKD sans PKRAD. Bien que les indications de la néphrectomie dans la PKRAD puissent être liées à la taille des reins, la décision de procéder à une néphrectomie native est souvent prise sur une base individuelle, sans référence spécifique aux mesures de la taille des reins.
Dialyse
Deux modalités de dialyse peuvent être utilisées dans le traitement des patients atteints de PKRAD : la dialyse péritonéale et l' hémodialyse . Les données épidémiologiques montrent que la PKRAD touche 5 à 13,4 % des patients hémodialysés en Europe et aux États-Unis, et environ 3 % au Japon. La dialyse péritonéale a généralement été contre-indiquée chez les patients atteints de PKRAD avec des volumes rénaux et hépatiques importants, en raison des difficultés physiques attendues lors de la procédure et des complications possibles ; cependant, aucune différence n'est observée dans la morbidité à long terme entre l'hémodialyse et la dialyse péritonéale dans la PKRAD.
Greffe du rein
La transplantation rénale est acceptée comme traitement de choix pour les patients atteints de PKRAD et d'IRT. Parmi les patients américains inscrits sur la liste d'attente d'une greffe de rein (en décembre 2011), 7 256 (8,4 %) étaient inscrits en raison d'une maladie kystique des reins et sur les 16 055 greffes rénales réalisées en 2011, 2 057 (12,8 %) l'ont été pour des patients atteints d'insuffisance kystique. néphropathie, dont 1189 de donneurs décédés et 868 de donneurs vivants.
Pronostic
Chez les patients atteints de PKRAD, le développement et l'expansion graduelles du kyste entraînent une hypertrophie rénale et, au cours de la maladie, le taux de filtration glomérulaire reste normal pendant des décennies avant que la fonction rénale ne commence à se détériorer progressivement, ce qui rend difficile la prédiction précoce de l'issue rénale. L'étude CRISP, mentionnée dans la section sur le traitement ci-dessus, a contribué à construire une justification solide soutenant la valeur pronostique du volume rénal total (TKV) dans la PKRAD ; Le TKV (évalué par IRM ) augmente régulièrement et un taux plus élevé d'hypertrophie rénale est corrélé à une diminution accélérée du DFG, tandis que le TKV ajusté en fonction de la taille du patient (HtTKV) ≥600 ml/m prédit le développement d'une maladie rénale chronique de stade 3 dans les 8 ans.
Outre TKV et HtTKV, le taux de filtration glomérulaire estimé (eGFR) a également été provisoirement utilisé pour prédire la progression de la PKRAD. Après l'analyse des tomodensitogrammes ou IRM de 590 patients atteints de PKRAD traités au Mayo Translational Polycystic Kidney Disease Center , Irazabal et ses collègues ont développé un système de classification basé sur l'imagerie pour prédire le taux de déclin de l'eGFR chez les patients atteints de PKRAD. Dans cette méthode pronostique, les patients sont divisés en cinq sous-classes de taux de croissance des reins estimés selon les plages de HtTKV spécifiques à l'âge (1A, <1,5 % ; 1B, 1,5 à 3,0 % ; 1C, 3,0 à 4,5 % ; 1D, 4,5 à 6,0 % et 1E, >6,0%) comme défini dans l'étude CRISP. Le déclin de l'eGFR au cours des années suivant la mesure initiale du TKV est significativement différent entre les cinq sous-classes de patients, ceux de la sous-classe 1E ayant le déclin le plus rapide. Certaines des causes de décès les plus courantes chez les patients atteints de PKRAD sont diverses infections (25 %), une rupture d'anévrisme des baies (15 %) ou une maladie coronarienne/hypertensive (40 %).
Les références
Liens externes
- https://web.archive.org/web/20110608142128/http://kidney.niddk.nih.gov/kudiseases/pubs/polycystic/index.htm
- https://www.ncbi.nlm.nih.gov/disease/PKD.html
| Classification | |
|---|---|
| Ressources externes |