
Procédé Wacker - Wacker process

Le procédé Wacker ou procédé Hoechst-Wacker (du nom des sociétés chimiques du même nom) fait référence à l'oxydation de l' éthylène en acétaldéhyde en présence de chlorure de palladium(II) comme catalyseur . Cette réaction chimique a été l'une des premières catalyses homogènes avec la chimie des organopalladiums appliquée à l'échelle industrielle.
Histoire
La réaction de Wacker a été rapportée pour la première fois par Smidt et al.
Le développement du procédé chimique maintenant connu sous le nom de procédé Wacker a commencé en 1956 à Wacker Chemie . À l'époque, de nombreux composés industriels étaient fabriqués à partir d' acétylène , dérivé du carbure de calcium , une technologie coûteuse et peu respectueuse de l'environnement. La construction d'une nouvelle raffinerie de pétrole à Cologne par Esso à proximité d'un site Wacker, combinée à la prise de conscience que l' éthylène serait une matière première moins chère, a incité Wacker à étudier ses utilisations potentielles. Dans le cadre de l'effort de recherche qui a suivi, une réaction de l'éthylène et de l'oxygène sur du palladium sur du carbone dans une quête d' oxyde d'éthylène a mis en évidence de manière inattendue la formation d'acétaldéhyde (simplement basée sur l'odeur). Des recherches plus poussées sur cette conversion de l'éthylène en acétaldéhyde ont abouti à un brevet de 1957 décrivant une réaction en phase gazeuse utilisant un catalyseur hétérogène. Entre-temps, Hoechst AG a rejoint la course et, après un dépôt de brevet, a forcé Wacker à créer un partenariat appelé Aldehyd GmbH . Le processus hétérogène a finalement échoué en raison de l'inactivation du catalyseur et a été remplacé par le système homogène à base d'eau pour lequel une usine pilote était opérationnelle en 1958. Les problèmes avec la solution catalytique agressive ont été résolus en adoptant le titane (nouvellement disponible pour un usage industriel) comme matériau de construction pour les réacteurs et les pompes. Les usines de production ont été mises en service en 1960.
Mécanisme de réaction
Le mécanisme réactionnel du procédé industriel Wacker (oxydation des oléfines via le chlorure de palladium(II)) fait l'objet d'une attention particulière depuis plusieurs décennies. Certains aspects du mécanisme sont encore débattus. Une formulation moderne est décrite ci-dessous :
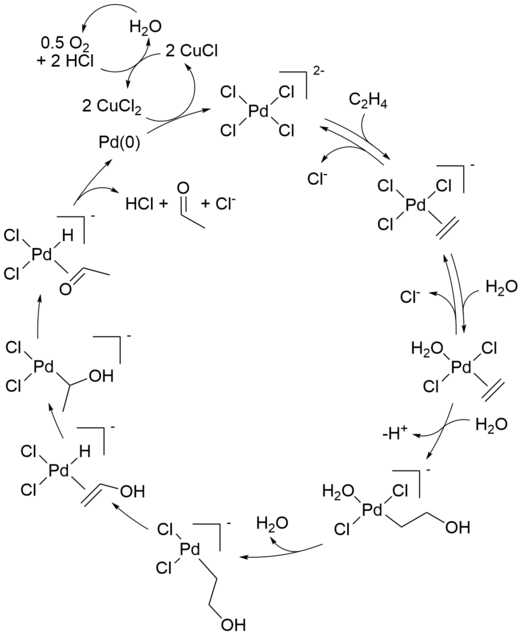
La réaction stoechiométrique initiale a été rapportée pour la première fois par Phillips. La réaction nette peut également être décrite comme suit :
- [PdCl 4 ] 2 − + C 2 H 4 + H 2 O → CH 3 CHO + Pd + 2 HCl + 2 Cl −
Cette conversion est suivie de réactions qui régénèrent le catalyseur Pd(II) :
- Pd + 2 CuCl 2 + 2 Cl − → [PdCl 4 ] 2− + 2 CuCl
- 2 CuCl + 1/2O 2 + 2 HCl → 2 CuCl 2 + H 2 O
Seuls l'alcène et l'oxygène sont consommés. Sans le chlorure de cuivre(II) comme agent oxydant , le métal Pd(0) (résultant de l' élimination du bêta-hydrure de Pd(II) dans l'étape finale) précipiterait, arrêtant la réaction après un cycle. Cette réaction stoechiométrique a été découverte en 1894. L'air, l'oxygène pur ou un certain nombre d'autres réactifs peuvent ensuite oxyder le mélange CuCl- chlorure résultant en CuCl 2 , permettant au cycle de continuer.
Études mécanistiques historiques
Les premières études mécanistes des années 1960 ont élucidé plusieurs points clés :
- Aucun effet d'échange H/D observé dans cette réaction. Des expériences utilisant du C 2 D 4 dans de l'eau génèrent du CD 3 CDO et des essais avec du C 2 H 4 dans du D 2 O génèrent du CH 3 CHO. Ainsi, la tautomérisation du céto-énol n'est pas une étape mécaniste possible.
- Effet isotopique cinétique négligeable avec des réactifs entièrement deutérés (k H/k D= 1,07). Par conséquent, on en déduit que le transfert d'hydrure n'est pas déterminant pour la vitesse .
- Effet isotopique compétitif significatif avec C 2 H 2 D 2 , (k H/k D= ~1,9), suggère que l'étape de détermination de la vitesse soit avant la formation d'acétaldéhyde.
- Des concentrations élevées de chlorure et de chlorure de cuivre(II) favorisent la formation d'un nouveau produit, la chlorhydrine .
De nombreuses études mécanistiques sur le procédé Wacker se sont concentrées sur la voie de formation de la liaison CO, l' étape d' hydroxypalladation . Henry a déduit que l'hydroxyde coordonné attaque le ligand éthylène, une voie interne (syn-). Plus tard, des études stéréochimiques menées par Stille et ses collègues soutiennent une voie anti-addition, par laquelle l'hydroxyde libre attaque le ligand éthylène. Les conditions des expériences de Stille diffèrent considérablement des conditions des procédés industriels. D'autres études utilisant des conditions industrielles normales de Wacker (sauf avec des concentrations élevées de chlorure et de chlorure de cuivre) ont également donné des produits qui ont déduit que l'attaque nucléophile était une réaction anti-addition.
Des études cinétiques ont été menées sur des alcools allyliques à substitution isotopique dans des conditions industrielles standard (avec de faibles concentrations de chlorure) pour sonder les mécanismes de réaction. Ces résultats ont montré que l'attaque nucléophile est un processus lent, tandis que les mécanismes proposés expliquant les études stéréochimiques antérieures supposaient que l'attaque nucléophile était un processus rapide.
Des études stéréochimiques ultérieures ont indiqué que les deux voies se produisent et dépendent des concentrations de chlorure. Cependant, ces études sont également contestées car les alcools allyliques peuvent être sensibles aux réactions d'isomérisation, et différents stéréoisomères peuvent être formés à partir de ces réactions et non à partir du procédé Wacker standard.
En résumé, les preuves expérimentales semblent soutenir que la syn-addition se produit sous des concentrations réactionnelles à faible teneur en chlorure (< 1 mol / l , conditions de procédé industriel), tandis que l'anti-addition se produit sous des concentrations réactionnelles élevées en chlorure (> 3 mol/l), probablement en raison des ions chlorure saturant le catalyseur et inhibant le mécanisme de la sphère interne. Cependant, la voie exacte et la raison de ce changement de voie sont encore inconnues.
Les questions sur le rôle du chlorure de cuivre compliquent encore le mécanisme du procédé Wacker. La plupart des théories supposent que le cuivre ne joue pas de rôle dans les mécanismes d'oxydation des oléfines. Pourtant, les expériences de Stangl et Jira ont révélé que la formation de chlorhydrine dépendait des concentrations de chlorure de cuivre. Les travaux de Hosokawa et de ses collègues ont donné un produit cristallisé contenant du chlorure de cuivre, indiquant qu'il pourrait avoir un rôle non innocent dans l'oxydation des oléfines. Enfin, une étude ab initio de Comas-Vives et al. impliquant aucun co-catalyseur de cuivre trouvé anti-addition était la voie préférée. Cette voie a ensuite été confirmée par des expériences sans cuivre par Anderson et Sigman. Une loi de vitesse cinétique différente sans dépendance aux protons a été trouvée dans des conditions sans cuivre, indiquant la possibilité que même de petites quantités de co-catalyseurs de cuivre puissent avoir des rôles non innocents sur cette chimie. Bien que ces travaux compliquent l'image du mécanisme du processus Wacker, on devrait probablement en déduire que cette chimie et la chimie connexe peuvent être sensibles aux conditions de réaction, et que plusieurs voies de réaction différentes peuvent être en jeu.
Une autre étape clé du procédé Wacker est la migration de l'hydrogène de l'oxygène vers le chlorure et la formation de la double liaison CO. On pense généralement que cette étape passe par une élimination dite d' hydrure avec un état de transition cyclique à quatre chaînons :

Des études in silico soutiennent que l' état de transition pour cette étape de réaction est défavorable et qu'un mécanisme alternatif de réaction d' élimination réductrice est en jeu. Les étapes de réaction proposées sont probablement assistées par une molécule d'eau en solution agissant comme un catalyseur.

Processus industriel
Deux voies sont commercialisées pour la production d'acétaldéhyde : un procédé en une étape et en deux étapes.
Processus en une étape
L'éthylène et l' oxygène sont passés à co- courant dans une tour de réaction à environ 130 °C et 400 kPa. Le catalyseur est une solution aqueuse de PdCl 2 et de CuCl 2 . L'acétaldéhyde est purifié par distillation extractive suivie d' une distillation fractionnée . La distillation extractive avec de l'eau élimine les pointes légères ayant des points d'ébullition inférieurs à ceux de l'acétaldéhyde ( chlorométhane , chloroéthane et dioxyde de carbone ) au sommet, tandis que l'eau et les sous-produits à point d'ébullition plus élevé, tels que l'acide acétique , le crotonaldéhyde ou les acétaldéhydes chlorés, sont extraits avec l'acétaldéhyde au fond. En raison de la nature corrosive du catalyseur, le réacteur est revêtu d'un matériau céramique résistant aux acides et le tube est en titane .
Processus en deux étapes
Dans le procédé en deux étapes, la réaction et l' oxydation sont effectuées séparément dans des réacteurs tubulaires. Contrairement au processus en une étape, l'air peut être utilisé à la place de l'oxygène. L'éthylène est passé à travers le réacteur avec le catalyseur à 105–110 °C et 900–1000 kPa. La solution de catalyseur contenant de l'acétaldéhyde est séparée par distillation éclair . Le catalyseur est oxydé dans le réacteur d'oxydation à 1000 kPa en utilisant de l'air comme milieu oxydant. La solution de catalyseur oxydé est séparée et renvoyée au réacteur. L'oxygène de l'air est complètement épuisé et l'air évacué est mis en circulation sous forme de gaz inerte. Le mélange acétaldéhyde-vapeur d'eau est préconcentré à 60 à 90 % d'acétaldéhyde en utilisant la chaleur de réaction et l'eau évacuée est renvoyée dans la tour éclair pour maintenir la concentration du catalyseur. Suit une distillation en deux étapes de l'acétaldéhyde brut. Dans la première étape, les substances à bas point d'ébullition, telles que le chlorométhane , le chloroéthane et le dioxyde de carbone , sont séparées. Dans la deuxième étape, l'eau et les sous-produits à point d'ébullition plus élevé, tels que les acétaldéhydes chlorés et l'acide acétique , sont éliminés et l'acétaldéhyde est obtenu sous forme pure en tête. En raison de la nature corrosive du catalyseur, les équipements en contact avec celui-ci sont revêtus de titane .
Dans les procédés à une et deux étapes, le rendement en acétaldéhyde est d'environ 95 % et les coûts de production sont pratiquement les mêmes. L'avantage d'utiliser des gaz dilués dans la méthode en deux étapes est compensé par des coûts d'investissement plus élevés. Les deux méthodes produisent des hydrocarbures chlorés, des acétaldéhydes chlorés et de l'acide acétique comme sous-produits. Généralement, le choix de la méthode est dicté par la situation des matières premières et de l'énergie ainsi que par la disponibilité de l'oxygène à un prix raisonnable. En général, 100 parties d'éthylène donnent :
- 95 parties d'acétaldéhyde
- 1,9 partie d'aldéhydes chlorés
- 1,1 partie d'éthène non converti
- 0,8 partie de dioxyde de carbone
- 0,7 partie d'acide acétique
- 0,1 partie de chlorométhane
- 0,1 partie de chlorure d'éthyle
- 0,3 partie d'éthane, de méthane, de crotonaldéhyde
et autres produits secondaires mineurs

Un organigramme montrant l'organigramme du processus Wacker en une étape pour la fabrication d'acétaldéhyde.

Un organigramme montrant l'organigramme du processus Wacker en deux étapes pour la fabrication d'acétaldéhyde.


Oxydation Tsuji-Wacker
L'avènement du procédé Wacker a stimulé de nombreuses recherches sur l'utilité et l'applicabilité des réactions à des oléfines terminales plus complexes. L' oxydation de Tsuji-Wacker est la transformation catalysée par le palladium (II) de ces oléfines en composés carbonylés. Clement et Selwitz ont été les premiers à découvrir que l'utilisation d'un DMF aqueux comme solvant permettait l'oxydation du 1-dodécène en 2-dodécanone, ce qui résolvait le problème d'insolubilité des oléfines d'ordre supérieur dans l'eau. Fahey a noté que l'utilisation de 3-méthylsulfolane à la place du DMF comme solvant augmentait le rendement d'oxydation du 3,3-diméthylbut-1-ène. Deux ans plus tard, Tsuji a appliqué les conditions de Selwitz pour les oxydations sélectives d'oléfines terminales avec de multiples groupes fonctionnels et a démontré son utilité dans la synthèse de substrats complexes. Le développement ultérieur de la réaction a conduit à divers systèmes catalytiques pour traiter la sélectivité de la réaction, ainsi qu'à l'introduction d'oxydations intermoléculaires et intramoléculaires avec des nucléophiles non aqueux.
Régiosélectivité
Ajout de Markovnikov
L'oxydation de Tsuji-Wacker oxyde l'oléfine terminale en la méthylcétone correspondante dans les conditions du procédé Wacker. Presque identique à celui du procédé Wacker, le cycle catalytique proposé (figure 1) commence par la complexation de PdCl 2 et de deux anions chlorure en PdCl 4 , qui subit ensuite l' échange de ligands de deux ligands chlorure pour l'eau et l'alcène pour former Pd (Cl 2 )(H 2 O)(alcène) complexe. Une molécule d'eau attaque ensuite l'oléfine de manière régiosélective via un mécanisme de sphère externe à la manière de Markovnikov , pour former le complexe Pd(Cl 2 )(OH)(-CH 2 -CHOH-R) thermodynamiquement plus stable . La dissociation d'un ligand chlorure aux trois complexes de palladium coordonnés favorise l'élimination de l'-hydrure, puis l' insertion migratoire subséquente de l'hydrure 1,2 génère le complexe Pd(Cl 2 )(OH)(-CHOHR-CH 3 ). Celui-ci subit une élimination de l' -hydrure pour libérer la cétone, et l'élimination réductrice subséquente produit du HCl, de l'eau et du palladium (0). Enfin, le palladium(0) est réoxydé en PdCl 2 avec deux équivalents de Cu(II)Cl 2 , qui à son tour peut être réoxydé par O 2 .
L'oxydation des oléfines terminales fournit généralement le produit de cétone de Markovnikov, cependant dans les cas où le substrat favorise l'aldéhyde (discuté ci-dessous), différents ligands peuvent être utilisés pour renforcer la régiosélectivité de Markovnikov. L'utilisation de la spartéine comme ligand (Figure 2, A) favorise la nucléopalladation au niveau du carbone terminal pour minimiser l'interaction stérique entre le complexe de palladium et le substrat. Le catalyseur au palladium ligaturé Quinox est utilisé pour favoriser la formation de cétones lorsque le substrat contient un groupe directeur (Figure 2, B). Lorsqu'un tel substrat se lie au Pd(Quinox)(OOtBu), ce complexe est saturé de manière coordonnée, ce qui empêche la liaison du groupe directeur et entraîne la formation du produit de Markovnikov. L'efficacité de ce ligand est également attribuée à sa propriété électronique, où le TBHP anionique préfère lier le trans à l'oxazoline et l'oléfine coordonne le trans à la quinoléine.

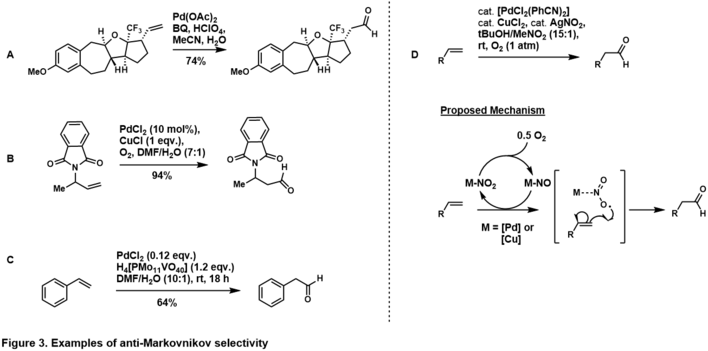
Ajout anti-Markovnikov
La sélectivité d'addition anti-Markovnikov à l'aldéhyde peut être obtenue en exploitant la stéréoélectronique inhérente au substrat. Mise en place du groupe diriger au homo-allyliques ( par exemple la figure 3, A) et allylique poste (Figure 3, B) à l'oléfine terminale favorise le produit aldéhydique anti-Markovnikov, ce qui suggère que , dans le cycle catalytique du groupe de direction chelates au complexe de palladium tel que l'eau attaque le carbone anti-Markovnikov pour générer le palladocycle plus stable thermodynamiquement. Une sélectivité anti-Markovnikov est également observée dans les substrats de styrényle (c'est-à-dire la figure 3, C), vraisemblablement via le complexe η 4 -palladium-styrène après que l'eau attaque l'anti-Markovnikov. D'autres exemples d'oxydation des oléfines anti-Markovnikov Tsuji-Wacker contrôlée par le substrat sont donnés dans les revues de Namboothiri, Feringa et Muzart.
Grubbs et ses collègues ont ouvert la voie à l'oxydation anti-Markovnikov des oléfines terminales stéréoélectroniquement impartiales, grâce à l'utilisation du système palladium-nitrite (Figure 2, D). Dans son système, l'oléfine terminale était oxydée en aldéhyde avec une sélectivité élevée via une voie de contrôle du catalyseur. Le mécanisme est à l'étude, mais des preuves suggèrent qu'il passe par un radical nitrite ajouté au carbone terminal pour générer le radical secondaire plus thermodynamiquement stable. Grubbs a étendu cette méthodologie à des oléfines plus complexes et impartiales.
Portée
Oxygène nucléophiles
Les oxydations intermoléculaires d'oléfines avec des alcools comme nucléophile génèrent typiquement des cétals , alors que les oxydations catalysées par palladium d'oléfines avec des acides carboxyliques comme nucléophiles genreats vinyliques ou allyliques carboxylates . Dans le cas des diols , leurs réactions avec les alcènes génèrent généralement des cétals, tandis que les réactions des oléfines portant des groupes électroattracteurs ont tendance à former des acétals .
Les oxydations intermoléculaires catalysées par le palladium de diènes avec des acides carboxyliques et des alcools comme donneurs donnent des produits d' addition 1,4 . Dans le cas du cyclohexadiène (figure 4, A), Backvall a constaté que le résultat stéréochimique du produit dépendait de la concentration de LiCl. Cette réaction se déroule en générant d'abord le complexe Pd(OAc)(benzoquinone)(allyle), par anti-nucléopalladation du diène avec de l'acétate comme nucléophile. L'absence de LiCl induit une élimination réductrice de la sphère interne pour permettre la stéréochimie du trans-acétate pour donner le trans-1,4-adduit. La présence de LiCl déplace l'acétate avec le chlorure en raison de son affinité de liaison plus élevée, qui force une attaque de l'acétate de la sphère externe contre le palladium, et permet à la stéréochimie cis-acétate de donner le cis-1,4-adduit. cyclisation oxydante intramoléculaire : le 2-(2-cyclohexényl)phénol se cyclise en dihydro-benzofurane correspondant (figure 4, B); L'acide 1-cyclohexadiène-acétique en présence d'acide acétique se cyclise en adduit lactone-acétate 1,4 correspondant (Figure 4, C), avec une sélectivité cis et trans contrôlée par la présence de LiCl.

Nucléophiles d'azote
Les aminations oxydantes des oléfines sont généralement conduites avec des amides ou des imides ; on pense que les amines sont protonées par le milieu acide ou qu'elles se lient trop étroitement au centre métallique pour permettre à la chimie catalytique de se produire. Ces nucléophiles azotés se révèlent compétents dans les réactions intermoléculaires et intramoléculaires, quelques exemples sont illustrés (Figure 5, A, B)
